Screening metabolico neonatale
Una novità per i genitori, disponibile già da qualche mese presso il Bioscience Institute, è la possibilità di effettuare al neonato uno screening completo sulle malattie metaboliche. Esistono malattie genetiche rare, in prevalenza metaboliche, non facilmente diagnosticabili; si manifestano nei primi giorni di vita con una sintomatologia generica che spesso viene confusa con quella di malattie più comuni. Lo screening metabolico neonatale ha lo scopo di diagnosticare precocemente tali malattie per stabilire una terapia efficace prima che possano causare gravi danni al bambino, soprattutto a livello neurologico. In molte nazioni del mondo sono stati avviati programmi obbligatori per lo screening neonatale delle malattie metaboliche. In Italia è obbligatorio solo per 4 malattie ereditarie. Il prelievo di alcune gocce di sangue deve essere effettuato non prima di 48 ore dalla nascita e non comporta rischi per il bimbo. Solitamente si consiglia di effettuarlo in concomitanza ai prelievi obbligatori per legge. Il sangue prelevato dal tallone del neonato viene disposto su un apposito cartoncino assorbente (test di Guthrie) e, una volta asciugato, viene inviato presso i laboratori della PerkingElmer Genetics che effettueranno delicate analisi. «Questa pratica – afferma il dottor Massimo Cervellini del Bioscience Institute – messa a disposizione gratuitamente ai genitori che si rivolgeranno al centro sammarinese, ha permesso di individuare già tre positività nei primi 5 mesi di operatività del servizio. Una patologia metabolica non diagnosticata può avere anche conseguenze letali per il neonato». Le malattie metaboliche ereditarie (MME) sono malattie genetiche rare: un errore nel DNA influenza negativamente l’attività enzimatica, a volte annullandola del tutto. Quando un enzima non funziona, influisce su una serie di reazioni chimiche essenziali per il funzionamento della cellula, degli organi e di conseguenza dell’organismo. Il gruppo di enzimi che lavora nelle reazioni chimiche necessarie per l’ossidazione dei nutrienti (zuccheri, proteine, grassi) riveste in tal senso una grandissima importanza. Le medesime reazioni sono fondamentali per fronteggiare il digiuno e trasformare le sostanze di riserva in energia. I difetti enzimatici che caratterizzano le MME sono pericolosi perché determinano effetti tossici per l’organismo nel caso in cui si assumano alimenti comuni (anche latte materno), oppure a seguito di un digiuno prolungato. Il sistema nervoso è il primo apparato a subire danni, spesso irreversibili. Se la malattia viene tempestivamente diagnosticata, tuttavia, è possibile evitare che l’intossicazione provochi danni permanenti al sistema nervoso attraverso una terapia alimentare e farmacologica mirata. La diagnosi di queste malattie è spesso tardiva perché la sintomatologia, piuttosto generica, compare nei primi giorni di vita e viene attribuita a patologie molto più comuni. La diagnosi delle MME può essere fatta solo attraverso analisi biochimiche, che non rientrano nella diagnostica di routine. Alcune forme intermedie e lievi si manifestano nel lattante o nel bambino; i primi sintomi si presentano allo svezzamento, o durante infezioni febbrili o nei casi di scarsa alimentazione. In altri casi la sintomatologia è caratterizzata da un ritardo di sviluppo o con la perdita delle capacità acquisite. Metodiche StepOne Tandem – mass La spettrometria tandem-mass è una metodologia diagnostica che permette di misurare, da una goccia di sangue, le sostanze presenti all’interno delle cellule durante le reazioni chimiche dovute all’ossidazione dei nutrienti. In un paziente affetto da deficit enzimatici si possono verificare delle alterazioni dal punto di vista delle reazioni chimiche, la cui conseguenza può essere l’eccesso di sostanza tossica o l’assenza di una sostanza necessaria per l’organismo. La PerkingElmer Genetics attraverso la spettrometria tandem-mass individua e misura quasi 60 sostanze, definite metaboliti, e valuta quindi, l’eventuale presenza di malattie metaboliche ereditarie.”¨ Analisi Biochimiche Spesso i disordini metabolici derivano da una mancanza o dal cattivo funzionamento di un enzima. In entrambi i casi l’organismo non produce i normali processi enzimatici. Attraverso le analisi biochimiche è possibile valutare tali disordini metabolici e quindi, le seguenti malattie:”¨ Fibrosi Cistica Galattosemia”¨”¨Ipotiroidismo Congenito”¨”¨Iperplasia Congenita In caso di screening positivo PerkinElmer Genetics, nel caso in cui i risultati allo screening risultino positivi, effettua approfondimenti diagnostici mediante analisi biomolecolari su campione. Frequentemente ad un disordine metabolico corrisponde un’alterazione di carattere genetico, causata quasi sempre da mutazioni genetiche. PerkinElmer Genetics, grazie all’impiego delle più sofisticate tecnologie diagnostiche è in grado di effettuare in tempi brevissimi le suddette analisi. Elenco delle Malattie Metaboliche Ereditarie StepOne A. Disordiniacidiorganici
- HMG
- Acidemia Glutarica Tipo I (GA I)
- Deficienza di Isobutiril-CoA Deidrogenasi
- Acidemia Isovalerica (IVA)
- Deficienza di 2-Methylbutryl-CoA Deidrogenasi
- Deficienza di 3-methylcrotonyl-CoA Carbossilasi
- Deficienza di 3-Methylglutaconyl-CoA Idratasi
- Acidemie Metilmaloniche
- Deficienza di Metilmalonil-CoA Mutasi
- Difetti sulla sintesi della adenosilcobalamina
- Deficienza di Vitamina B12 materna
- Deficienza di Acetoacetil-CoA Tiolasi Mitocondriale
- Acidemia Propionica
- Deficienza multipla di (CoA) Carbossilasi
- Aciduria Malonica
B. Disordininell’OssidazionedegliAcidi
- VLCAD ( Very Long Chain Acyl-CoA Dehydrogenase Deficiency)
- Deficienza di Carnitinia/Acilcarnitina translocasi
- Deficienza di Carnicina Transferasi Tipo I
- MCAD ( Medium chain Acyl-CoA Dehydrogenase Deficiency)
- LCHAD (3-Hydroxy Long Chain Acyl-CoA deydrogenase Deficiency)
- Deficienza di 2,4-Dienol-CoA Reduttasi
- MAD ( Multiple Acyl-CoA Dehydrogenase Deficiency )
- CPT Tipo II ( Neonatalo Carnicina Palmitoyl Transferase Deficiency )
- SCAD (Short Chain Acyl-CoA Dehydrogenase Deficiency)
- SCHAD (Short Chain Hydroxy Acyl-CoA Dehydrogenase Deficiency )
- Deficienza TFP ( Trifunctional Protein Deficiency)
C. Disordini negli Aminoacidi
- Argininemia
- Aciduria Argininosuccinica
- 5-Ossoprolinuria
- Deficienza di carbamoilfosfato Sintetase
- Ctrullinemia
- Omocistinuria
- Ipermethioninemia
- Iperammonemia, Iperornithinemia, Omocitrullinemia ( Sindrome )
- Iperornitinrmis con Atrofia
- Malattia “ Maple Syrup Urine”
- Fenilchetonuria:
- Classica-Iperfenilalaninemia
- Cofattore Biopertina
- Tirosinemia:
- Tirosinemia Transiente Neonatale
- Tirosinemia Tipo I
- Tirosinemia Tito II
- Tirosinemia Tipo III
Patologie analizzate con tecniche biochimiche e biomolecolari:
- Deficienza di Biotinidasi:
- Completa
- Parziale
- Iperplasia Congenita Adrenale:
- Deficienza di 21-idrossilasi “ Salt Wasting”
- Deficienza di 21-idrossilasi “ Simple Virilizing”
- Ipotiroidismo Congenito
- Fibrosi Cistica
- Galattosemia:
- Deficienza di Galattocinasi
- Deficienza di Galattosi-1-fosfato Uridiltransferasi
- Deficienza di Galattosi-4-Empimerasi
- Deficienza di Glucosio-6-Fosfato Deidrogenasi
- Emoglobinopatie
- Emoglobina S, S/C, S/Beta-Talassemia, Malattie C, & E
Altre analisi:
- Iperalimentazione
- Disturbi del fegato
- Gestione dei trigliceridi a media catena
- Presenza di EDTA Anticoaugulanti nel campione di sangue
- Trattamento con Benzoato, Acido Rivalico, Acido Valproico
- Deficienza di Assorbimento di Carnicina
Potrebbe interessarti

Dispositivo di monitoraggio neonatale
Un gruppo di ingegneri della General Electric ha creato un dispositivo per monitorare la respirazione dei neonati nati prematuramente. E’ noto che i bimbi prematuri vanno spesso incontro a crisi respiratorie e sempre devono trascorre un periodo in incubatrice, il nuovo marchingegno sarà in grado di monitorare non soltanto l’attività respiratoria ma anche quella cardiaca.… Continua a leggere Dispositivo di monitoraggio neonatale
Indonesia: neonato da Guinnes dei primati
Il neonato piu’ grande del mondo pesa più di 8 chili. Il piccolo è nato proprio in questi giorni in Indonesia ed è già una star. Suo il record per il neonato più grande. Il bambino non ha ancora un nome. A darlo alla luce la madre Ani, di 41 anni, in un ospedale di Kisaran, a nord dell’Isola… Continua a leggere Indonesia: neonato da Guinnes dei primati
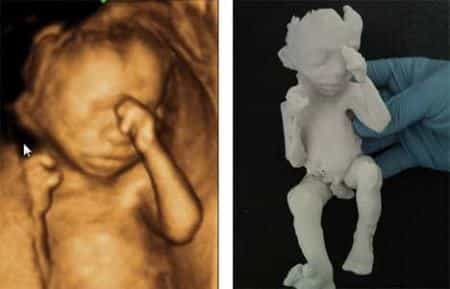
Prototipazione rapida del nascituro
Una delle più belle esperienze per un genitore è osservare il neonato in pancia grazie a tecniche a ultrasuoni, ecografia in primis, meglio se di ultima generazione con la possibilità di visualizzare immagini 3D e 4D. Gli studenti del Royal College of Art design hanno fatto un ulteriore passo per far “toccare con mano” il… Continua a leggere Prototipazione rapida del nascituro
Morte in culla – novità
La sindrome della morte improvvisa del lattante (Sudden Infant Death Syndrome- SIDS) è un evento tragico che nei paesi occidentali rappresenta ancora la principale causa di morte nel primo anno di età . La concomitanza di fattori di rischio genetici ed ambientali rende particolarmente difficile individuarne le cause. Uno studio pubblicato su Science offre ora importanti… Continua a leggere Morte in culla – novità
IBM in aiuto ai nati prematuramente
IBM sta attivamente collaborando con lo University of Ontario Institute of Technology per produrre un software in grado di monitorare da vicino e prevedere lo stato di salute dei neonati messi al mondo troppo prematuramente. Le prime fasi sono quelle più critiche ed è proprio qui che si dovrebbe inserire questo analizzatore. Secondo alcuni test,… Continua a leggere IBM in aiuto ai nati prematuramente